
发布人:管理员 发布时间:2022-07-23
2.注册单元划分
2.1工作原理
应描述手术器械的工作原理。
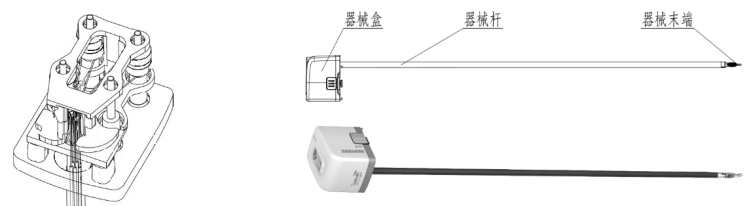
a.器械盒内部结构 b.手术器械整体图示
图1 手术器械结构

a.单极弯剪(含能量护套) b.单极电钩 c. 单极电铲
图2 单极手术器械举例

a.双极钳 b.双极镊
图3 双极手术器械举例

a.持针钳 b.手术剪 c.抓持器 d.施夹钳(含结扎夹)
图4 不向患者提供能量的手术器械举例
表1 手术器械列表举例
|
|
|
|
|
|
|
|
|
|
|
|
|
本指导原则范围内的手术器械对医疗器械安全和性能基本原则清单的符合性见附录2,注册申请人可参考《医疗器械安全和性能基本原则》符合性技术指南编写该清单。
3. 产品技术要求及检验报告
对于无菌提供的手术器械应提供货架有效期和包装研究资料,可通过对产品和包装进行加速老化和/或实时老化试验及运输试验,试验后对产品进行性能、功能和无菌检测,对包装外观、完整性和无菌屏障系统的性能进行验证,证明产品在货架有效期内保持无菌且性能功能符合预期要求。加速老化和无菌屏障系统的性能验证可参考YY/T 0681系列标准。
|
|
|
|
|
|
|
分)与带电部分隔离/保护不够,可能引起过量漏电流对操作者或患者造成电击危害。 |
|
|
||
|
|
||
|
|
||
|
|
||
|
|
|
|
|
|
||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
|
|
|
|
|
|
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
|
|
|
|
||
|
|
||
|
|
||
|
|
|
|
|
|
||
|
|
||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
标准编号 |
标准名称 |
|
GB 9706.1 |
医用电气设备 第1部分:安全通用要求 |
|
GB 9706.1 |
医用电气设备 第1部分:基本安全和基本性能的通用要求 |
|
GB 9706.4 |
医用电气设备 第2-2部分:高频手术设备安全专用要求 |
|
GB 9706.202 |
医用电气设备 第2-2部分:高频手术设备及高频附件的基本安全和基本性能专用要求 |
|
GB 9706.19 |
医用电气设备 第2部分:内窥镜设备安全专用要求 |
|
GB 9706.218 |
医用电气设备 第2-18部分:内窥镜设备的基本安全和基本性能专用要求 |
|
YY 9706.277 |
医用电气设备 第2-77部分:采用机器人技术的辅助手术设备基本安全和基本性能的专用要求 |
|
YY 0505 |
医用电气设备 第1-2部分:安全通用要求 并列标准:电磁兼容 要求和试验 |
|
YY 9706.102 |
医用电气设备 第1-2部分:基本安全和基本性能的通用要求 并列标准:电磁兼容 要求和试验 |
|
YY/T 0686 |
医用镊 |
|
YY/T 0940 |
医用内窥镜 内窥镜器械 抓取钳 |
|
YY/T 0941 |
医用内窥镜 内窥镜器械 咬切钳 |
|
YY/T 0943 |
医用内窥镜 内窥镜器械 持针钳 |
|
YY/T 0944 |
医用内窥镜 内窥镜器械 分离钳 |
|
YY 0672.2 |
内镜器械 第2部分 腹腔镜用剪 |
|
YY/T 1686 |
采用机器人技术的医用电气设备 分类 |
|
YY/T 1712 |
采用机器人技术的辅助手术设备和辅助手术系统 |
附录4:产品技术要求中手术器械性能指标

 官网关注
官网关注 0769-83110798
0769-83110798 E-mail
E-mail